立院三讀通過
醫療器材管理法,加速醫療器材上市
WHY : 衛福部在醫療器材管理法的規定下
1. 依照產品風險程度分類及分級管理
2. 國內醫療器材製造商多元經營模式下與藥品商有差異
3. 國內與國際間醫療器材管理規範接軌
醫療器材管理法將由藥事法中獨立出來,在未來藥事法有關醫療器材之相關規定將不 再適用。
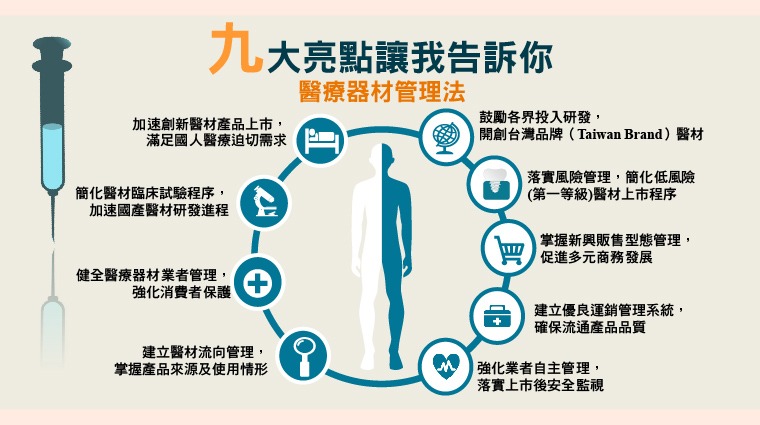
增加醫療器材業者適用範圍,泛指醫療器材製造商與銷售商,
列指這兩類:
1. 從事醫療器材製造、包裝、標籤、滅菌與最後檢驗放行
2. 從事醫療器材之開發、並以其開發設計名義於市場流通
經中央機關公告-風險等級之醫療器材,製造商及醫事機構應該建立保存商品直接供應 來源之流向資料。
針對低風險醫療器材由查驗登記制度改由電子式線上登錄,且以年度申報登陸保持有 效性。
為了落實風險管理,經公告無風險之醫療器材臨床實驗不需申請主管機關核准 臨床實驗若試驗者發生:死亡、危害性命、暫時或永久性失能、試驗者之胎兒(嬰兒)先 天性畸形、需住院或延長住院或其他可能導致永久性傷害之併發症,之任何情況下, 皆應通報。
強化上市後醫療器材之安全監督管理: 醫療器材供應商依照公告或核定安全監督計畫期安全性;醫療機構須協助提供相關監 督資料予醫療器材供應商。
醫療器材供應商需將安全監督報告定期提供中央主管機關,若中央主管機關認定該產 品有安全及監督計畫內容與公告規定不符合之疑慮時將其令其暫停製造、輸入販售等 視情況重大者將予終止許可證之核發或登錄。
以上敘述安全監督資料報告評估與其他相關事項辦法及實施規則,全由中央主管機關 規定。
導入 ISO13485 之相關業者,應該注意法規最新動向,以隨時調整管理系統符合。
免責聲明
轉載此文是出於傳遞更多訊息為目的,若有來源標註錯誤或侵犯了您的合法權益,請作者持權屬證明與本網聯繫,我們將及時更正、刪除,謝謝。
參考資料:
https://www.mohw.gov.tw/cp-16-50552-1.html
 Home
Home
 TOP
TOP